
Background
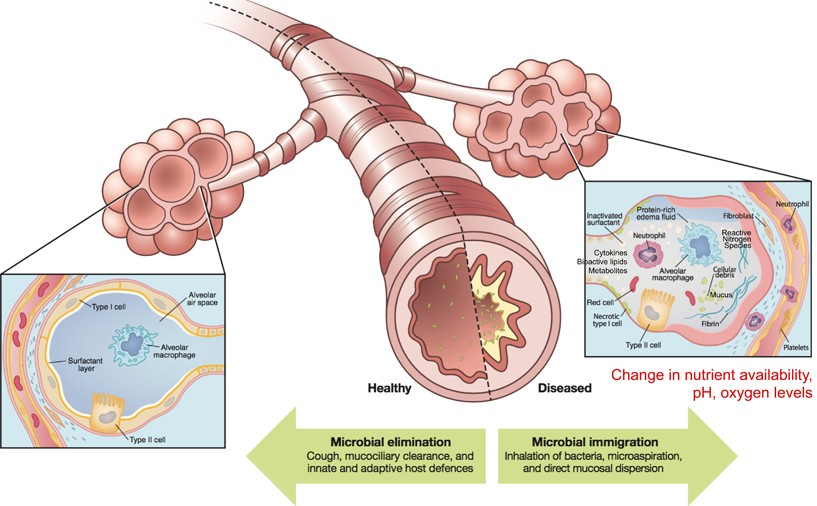
Monogenic disorders are estimated to affect 1/100 people at birth (WHO). They are caused by individual mutations in individual genes that result in non-functional products such as RNA molecules or proteins. In the case of cystic fibrosis (CF), a mutation in an ion-transport protein causes CF patients to have thick, sticky mucus that easily traps bacteria, viruses and other contaminants that can cause disease. Thick mucus is particularly an issue in the lungs where trapped microbes can lead to lung inflammation and infection (Cystic Fibrosis Foundation). In healthy individuals, microbes are not uncommon in the upper and lower respiratory tracts. Usually, microbes are eliminated by little hairs that sweep microbes trapped in free-flowing mucus up and out of the respiratory tract to be coughed up or swallowed (Huffnagle et al., 2016). However, in CF patients thick mucus limits this elimination of microbes, trapping them in the lungs and respiratory tract (Fig. 1).
Continue reading “The changing microbiome in cystic fibrosis: a key to diagnosis and treatment?”